Protocol article / Open Access
DOI: 10.31488/bjcr.195
A phase 2 Double-Blind, RandomizEd, Prospective, Placebo Controlled Study of NanO2 TM Combined With Radiation and Temozolomide in Patients with Newly-Diagnosed Glioblastoma MultiformE: RESTORE
Jennifer LH Johnson, PhD*1, Evan Unger, MD2
1. CSO of NuvOx Pharma, USA
2. CEO of NuvOx Pharma, USA
*Corresponding author: Jennifer LH Johnson, CSO of NuvOx Pharma, 1635 East 18th Street, Tucson, Arizona, USA 85719
Abstract
This article details the protocol of a Phase 2 clinical trial treating a specific type of brain cancer (glioblastoma). The name of the trial is: A phase 2 double-blind, RandomizEd, prospective, placebo-controlled STudy of NanO2TM combined with radiation and temozolomide in patients with newly-diagnosed glioblastoma multiformE: RESTORE. Eighty-seven glioblastoma patients are being recruited and randomized to supplement the standard of care with either study drug (NanO2™) or placebo at a ratio of 2 to 1. The primary outcome is progression free survival and the secondary outcome is overall survival. To date, 11 patients have been treated without any drug related adverse effects. The study is ongoing and expected to be completed by the 3rd quarter of 2025.
Keywords: Hypoxia, oxygen delivery, glioblastoma, clinical trial, protocol, chemoradiation, dodecafluoropentane, perfluoropentane, brain tumor
Introduction
Glioblastoma multiforme
Glioblastoma multiforme (GBM) is the most common primary malignant tumor of the central nervous system (CNS), with approximately 10,000 new cases diagnosed annually in the USA [1]. Despite initial treatment with surgery, radiation therapy and chemotherapy, most patients develop recurrent disease and only approximately 25% survive 2 years [2]. Standard therapeutic options for recurrent or progressive GBM are associated with limited efficacy. Second-line chemotherapy results in low objective response rates and progression-free survival at 6 months (PFS-6) is typically less than 20% [3]. Anti-angiogenic agents such as bevacizumab are associated with higher response rates, but durable responses are rare [4]. Clearly, better therapies for GBM are urgently needed.
Rationale for targeting tumor hypoxia in GBM
Glioblastomas typically contain significant regions of hypoxic tumor tissue. The cause of the hypoxia is uncertain, but it might arise from thrombosis of small blood vessels due to production of pro-coagulant factors by the tumor cells [5, 6]. Hypoxia leads to necrosis of parts of the GBM and also the switching on of expression of angiogenic factors, most notably vascular endothelial growth factor (VEGF) [7-10]. One important consequence of tumor hypoxia is relative resistance to therapeutic radiation. This occurs because radiation-induced killing of malignant cells depends on the production of oxygen free radicals by the ionizing radiation [11]. If the tumor has hypoxic regions, oxygen free radicals will form poorly after irradiation and the tumor cells will be resistant to treatment. Similarly, hypoxic cells can become chemoresistant due to decreases in drug action in the absence of O2, limited drug diffusion, and inability to deliver to cells distant from functional vasculature [12]. The hypothesis of this investigation is that increasing oxygen delivery to hypoxic GBM tissue will boost the effectiveness of frontline treatment with both radiation therapy (RT) and chemotherapy (CT).
In recent years, the causal relationship between oxygen and radiation response in hypoxic tumors has become a well-known paradigm. The radiosensitization results from the increased levels of molecular oxygen, which leads to chemical reactions that produce DNA damage after the absorption of energy from ionizing radiation [13]. One of the major obstacles is the lack of vasculature in hypoxic tumors, which prevents high levels of oxygen from reaching the hypoxic areas of the tumor. A method to increase delivery of oxygen to the anoxic cells will better reverse the effects of tumor hypoxia, thereby improving the clinical outcome through increased radiosensitization.
The relationship between oxygen and chemotherapeutics is a less studied area of research, most likely due to the fact that the effect of oxygen is specific to the mechanism of action of the drug. A recent, in vitro, study demonstrated the effects of hyperoxia on human glioblastoma cells resistant to temozolomide (TMZ) [14]. TMZ-sensitive GBM cells were repetitively exposed to TMZ to develop subclones of TMZ-resistant GBM cells. These TMZ-resistant cells were then exposed to varying oxygen levels, with or without TMZ treatment. The results showed that the TMZ sensitivity of both chemo-sensitive and resistant cells was significantly increased under hyperoxia. The specific mechanism of hyperoxia to the enhanced TMZ toxicity was hypothesized to be due to the induction of apoptosis, specifically via MAPK-related pathways [14]. This study suggests that increased oxygen delivery may potentially result in an improved clinical outcome in both TMZ responders and non-responders.
There have been various modalities tested to increase oxygen delivery to hypoxic tumors in order to increase response to therapy. Hyperbaric oxygen and carbogen (mixtures of oxygen with carbon dioxide, e.g., 95-98% O2 with 2-5% CO2) have been tested as a means of increasing tumor oxygen, but have not produced obvious improvements in clinical outcomes [15, 16]. The oxygen-carrying capacity of the blood is limited, and hemoglobin is quickly saturated by supplemental oxygen. Oxygen therapeutics (OTs) based on liquid fluorocarbons potentially can increase the oxygen-carrying capacity of blood; however, they required high doses of the fluorocarbons and were associated with adverse events in clinical trials. OTs based upon hemoglobin derivatives were also associated with adverse events and failed in clinical trials. Fluosol is an example of a liquid fluorocarbon that has been tested extensively for the treatment of GBM. Like other drug products from this class of therapeutic, Fluosol is administered at high doses, which leads to toxicity in patients. However, the results from the pre-clinical and clinical studies of Fluosol strongly relate to the predicted outcome of studies with NanO2 since both products act to deliver oxygen to hypoxic tissue with the same mechanism of action.
A preliminary study was done to analyze the OT, Fluosol-43, as a CT sensitizer using the chemotherapeutic agent, BCNU, on a rat brain model of glioma [17]. Fluosol-43, or perfluorotributylamine, had two times greater oxygen solubility than hemoglobin and was predicted to have a beneficial effect on hypoxic cells and/or poorly vascularized areas in malignant gliomas. The study was done in Wistar rats, in which glioma tumor cells were transplanted into the brain. After 10 days, the rats were treated with either Fluosol-43, BCNU, Fluosol-43 + BCNU, or no treatment (control). The results showed that the combined effect of Flluosol-43 plus BCNU in an oxygen environment produced a significant increase in mean survival time compared to that of the BCNU treatment alone. The synergistic effect of Fluosol with BCNU was hypothesized to be due to two major factors: 1. The high oxygen transport of Fluosol most likely oxygenated hypoxic cells 2. The addition of Fluosol-43 may have decreased the overall blood viscosity, increasing the cerebral blood flow, which thereby increased delivery of BCNU to the tumor [17]. We believe this study can translate to the use of NanO2 as a chemosensitizer to TMZ since the combined mechanism of action of Fluosol increasing tumor oxygenation to enhance the effects of BCNU is the same.
Fluosol is the only fluorocarbon emulsion besides NanO2 to be tested in clinical studies as a radiosensitizer. However, the doses of Fluosol required in the clinical trials were approximately 800 to 1600-fold higher than for NanO2, on a gram basis of fluorocarbon.
In summary, Fluosol has been studied as a radiosensitizer in several studies, including in association with hyperbaric oxygen chamber in GBM [22]. The doses of fluorocarbon (FC) used were about 800-1,600 times higher per dose than for the dose of FC in NanO2. Fluosol could only be administered a maximum of one time per week (unlike NanO2 which can be administered during each fraction of radiation – 5 times per week). While there have been minimal and no serious acute adverse events with administration of NanO2, Fluosol administration was associated with adverse events. In the largest study, 98 subjects with recurrent GBM, 68% of subjects had alterations of vital signs. Overall, allergic reactions were common, including altered liver enzymes, nausea, and vomiting as well as other adverse reactions. NanO2 is the first oxygen therapeutic capable of administration during each fraction of RT without causing significant adverse reactions. Current results from the Phase Ib study of NanO2 in GBM subjects indicate that there are minimal adverse events in subjects, and a trend of improvement in the overall survival of subjects.
NanO2: A High-Capacity Oxygen Carrier
NanO2 is an emulsion of 2% dodecafluoropentane (DDFP) in stabilizers (sucrose, PTB) and phosphate buffered saline pH 7.2. Compared to higher molecular weight liquid fluorocarbons that have already been studied extensively as OTs, dodecafluoropentane in NanO2 carries far more oxygen per gram of fluorocarbon. Consequently, if it were administered intravenously (IV) and passed through the lungs of subjects breathing high concentrations of oxygen, it would take up large amounts of oxygen and increase the oxygen concentration in blood in a dose-dependent way. The intent of this study is to demonstrate that by shifting hypoxic tumor environments to normal tissue pO2 environments, the effectiveness of radiation treatment will be improved.
Preclinical efficacy studies in cancer
In mice implanted with human pancreatic tumor xenografts, survival of the NanO2 treated group (NanO2 + RT + carbogen) was twice that of mice treated with radiation treatment alone (RT + carbogen) [23]. Tumor pO2 was measured in the pancreatic xenografts with an oxygen electrode. Compared to carbogen alone, NanO2 + carbogen resulted in a 400% increase in tumor oxygen levels. In another study of DDFPe (identical material to NanO2 but without a physiological buffer), administration with carbogen completely reversed radiation resistance [24]. Preliminary studies at the University of Arizona have demonstrated that there is no difference between carbogen and 100% oxygen with respect to oxygenation of tumors in mice [25]. Therefore, 100% oxygen is used within this protocol.
Non-clinical pharmacology and toxicology
Extensive work was done to evaluate the toxicity and PK/PD of NanO2 during its development as an ultrasound contrast-imaging agent by Sonus Pharmaceuticals.[1,2] During development, most work was done using "activated" NanO2 referred to as EchoGen® when it was studied as a contrast agent. EchoGen was administered as bolus IV injections. Activation was achieved by applying negative pressure (usually by suction in a syringe) which resulted in 2 micron sized microbubbles of gas in the circulation. In preclinical toxicity studies of EchoGen, it was found that the more efficient the activation method was in producing microbubbles, the lower the minimum lethal dose because an increased concentration of larger particles led to a greater chance of toxicity through pulmonary micro-embolism. The final method of activation, hypobaric activation, resulted in rat mortality at doses of 2.0 mL/kg, which is half the lethal dose using filter activation (a less efficient method at producing microbubbles). Therefore, the “inactivated” form of NanO2 (0.2 micron vesicles of NanO2 without microbubbles) is expected to have an improved safety profile. In a single-dose rat study, the No Observable Adverse Effect Level of “inactivated” NanO2 was 4.0 mL/kg. This converts to a human equivalent dose (HED) of approximately 0.64 mL/kg.[26, 27] At the higher dose level in rat study (6.0 mL/kg), toxicity was observed as impaired movement, labored breathing and lethargy. No mortality was observed, and all animals recovered within 24 hours.
Studies have also been performed in non-human primates. Anaesthetized rhesus monkeys received 0.4 mL/kg of EchoGen IV every 30 minutes for 3 doses without adverse effects. In addition, unconscious cynomolgus monkeys showed no adverse reactions to 0.4 mL/kg of EchoGen given IV every 30 minutes for 3 doses and showed only minor transient changes in blood pressure and heart rate at up to 1.1 mL/kg. Conscious cynomolgus monkeys received escalating doses of 0.05, 0.1, 0.2, 0.4 0.6 & 0.8 mL/kg 30 minutes apart. The NOAEL in this study was 0.6 mL/kg (or 1.35 mL/kg cumulative). On a single dose basis, the NOAEL of 0.6 mL/kg converts to an HED of 0.19 mL/kg.
Additional information on the preclinical evaluation of NanO2 is presented in the Investigator’s Brochure.
Pharmacokinetics and pharmacodynamics of NanO2 in human subjects
NanO2 was tested between 2017 and 2019 in an Australian phase Ib/II dose finding study of NanO2 combined with radiation and TMZ in patients with newly-diagnosed glioblastoma multiforme. All subjects received standard chemoradiation consisting of 30 fractions of focal brain radiation (total 60 Gray, given as 2 Gray fractions on 5 days per week for 6 weeks) with concurrent oral TMZ at a dose of 75 mg/m2 day on 7 days per week for 6 weeks. NanO2 was administered by IV infusion over 30 minutes in combination with each fraction of radiation. Subjects continuously breathed either 100% oxygen or carbogen (a mixture of oxygen 98% and carbon dioxide 2%) from the start of each NanO2 infusion.
A randomized, open-label, multi-dose Phase Ib/II study with NanO2 as a radiosensitizer in patients with glioblastoma multiforme brain tumors has been completed. The clinical study evaluated the safety and tolerability of NanO2 following IV infusion in post- surgical GBM patients undergoing fractionated RT with concomitant chemotherapy with temozolomide. A total of 11 patients were enrolled in the trial. In the first dose exploration phase, 3 patients were enrolled, one each in 0.05, 0.1 and 0.17 ml/kg dose cohorts. The MTD was defined as 0.1mL/kg in this 30-dose regimen as two patients treated at 0.1 and 0.17mL/kg (01-002 and 01-003 respectively) had late toxicity (increased radiation necrosis) considered possibly related to NanO2. Although these events occurred beyond the 6-week MTD period, it was decided to treat the radiation necrosis events as if they were DLTs for the safety of the patients. In the dose expansion phase, 8 additional patients were enrolled at 0.1mL/kg.
Seven of the eleven patients completed both the chemoradiation period and the adjuvant TMZ period of the study. The remaining four patients were withdrawn from the study, following completion of the chemoradiation period, but prior to completion of the adjuvant TMZ period. Reasons for withdrawal included 02-004 - Investigator’s decision, 02-007 - Disease progression, 03-008 - Death, and 01-009 - Adverse event.
One patient (01-003) receiving 0.17 mL/kg had decreased motor function in the right arm and was briefly hospitalized for these symptoms. At the time, it was felt by the investigators that these symptoms were not drug related but related to underlying disease and radiation therapy (RT). The patient discontinued NanO2 (received 28 out of 30 doses), however and finished radiation treatment. Six weeks after finishing RT the patient had headache and a repeat MRI scan showed enhancing tissue consistent with radiation necrosis. Surgery was performed confirming radiation necrosis. The degree of radiation necrosis which was observed was more extensive and earlier than normally expected. For this reason, it was determined that the earlier symptoms that the patient experienced may have been related to the increased oxygenation associated with NanO2, and therefore were determined to be a DLT. Additional patients were then enrolled at the recommended daily dose of 0.1 mL/kg dose.
Eight patients (73%) experienced at least one non-serious TEAE. The most prevalent non-serious TEAEs that were considered possibly, probably or definitely related to NanO2 were fatigue (six events, n=4, 36%), headache (two events, n=2, 18%), and decrease in platelet count (10 events, n=2, 18%). Five patients (46%) experienced a total of 9 serious TEAEs. A total of 3 serious TEAEs were considered possibly or probably related to NanO2, which included radiation necrosis, cognitive disorder and decrease in platelet count. Note that temozolomide chemotherapy is known to induce thrombocytopenia.[33] No acute toxicity due to injection or administration was observed from daily IV dosing Monday through Friday for 6 weeks at the 0.1 mL/kg dose nor from daily dosing at the 0.17 mL/kg level (delayed toxicity may have occurred at this dose level due to enhanced radiation necrosis).
According to review of the serial MR images, the median time to progression was 9.6 months (292 days) compared to a historical control of 6.9 months.[4] Overall survival was analyzed using Kaplan-Meier statistics at every 30 days from first dose of NanO2. There were six deaths confirmed as part of the study and its follow-up. The remaining five patients were classified as alive at the time of last contact. Historically, the average overall survival for GBM patients, for both unmethylated and methylated MGMT, is about 14.6 months [4]. The median overall survival for the study was 19.4 months (591 days).
Pharmacokinetic data collected during studies conducted by Sonus were analyzed by NuvOx.[2] The pharmacokinetics of DDFP in human subjects exhibits biphasic decline after an IV bolus dose; there is a rapid initial decline followed by a slow terminal elimination phase. The elimination half-life in humans ranges from 81-99.5 min for doses of 0.01-0.1 mL/kg. The values of terminal half-life in dogs (30-48 min) were shorter than humans. The terminal half-life is the shortest in rats (0.7-1.9 min). Efficacy was demonstrated in lower levels in animal stroke models and higher levels are not thought necessary in humans.
Sonus tested the safety of activated DDFPe as a contrast agent in patients undergoing echocardiography with Class III to IV congestive heart failure (CHF) or severe obstructive pulmonary disease (COPD). Two studies were performed comparing the safety of the drug to placebo saline. Activated DDFPe was administered as an injection of 0.08 mL/kg in 146 patients with CHF and 134 patients with COPD. There were no significant differences in adverse events in either the CHF or COPD patients of study drug compared to placebo.[35] Note that in studies as a contrast agent, most patients were dosed with 0.05 mL/kg as higher doses resulted in acoustic attenuation. The safety studies, in perhaps the most vulnerable patient population with CHF and COPD, shows safety at the level of 0.08 mL/kg from rapid IV bolus injection. In the RESTORE Trial, drug is not activated, i.e. injected as the neat emulsion with roughly 1/10th the particle diameter of the activated form, and also injected as slow IV push.
DDFP is not metabolized in humans. The major route of excretion of DDFP is through expired air. Almost one hundred percent of administered dose was recovered in the expired air as DDFP. The clearance of DDFP in humans is 43.9-65.2 mL/min/kg.
Rationale for Combining NanO2 with Chemoradiation as Frontline Therapy of GBM
NanO2 will be administered as an IV push concomitant with 100% oxygen in combination with each radiation fraction during radiation for newly-diagnosed GBM. The rationale is to increase the amount of oxygen being delivered in blood to the tumor and therefore to increase sensitivity to irradiation. Subjects will also receive a standard regimen of concurrent chemotherapy [2].
Materials and Methods
Study synopsis
Study title
A phase 2 double-blind, RandomizEd, prospective, placebo controlled STudy of NanO2TM combined with radiation and temozolomide in patients with newly-diagnosed glioblastoma multiformE: RESTORE
Primary objective
1. To determine progression free survival (PFS) in newly-diagnosed glioblastoma patients after treatment with NanO2 in combination with radiation and temozolomide
Secondary objectives
1. To determine overall survival after treatment with NanO2 in combination with radiation and temozolomide
2. To determine the objective response rate to study therapy using the modified Response Assessment in Neuro-oncology (mRANO) criteria
3. To determine the effect of NanO2 on the rate of pseudoprogression
4. To confirm that NanO2 re-oxygenates glioblastoma multiforme
5. To estimate the effect on the duration of functional independence as measured on the Karnofsky Performance Scale
6. To use questionnaires to study patient quality of life using FACT-Br
7. To use questionnaires to study caregiver quality of life using CQOLC
Exploratory objectives
1. To use MRI technologies to characterize the in-vivo effects of NanO2 on tumor oxygenation as a pharmacodynamics biomarker
2. To use FACT-Br and CQCOLC quality of life questionnaires to gain patient and caregiver perspectives for benefit/risk analysis
Study design
This randomized, prospective, placebo-controlled trial.
• All subjects will receive standard chemoradiation consisting of 30 fractions of focal brain radiation
• Total 60 Gy, given as 2 Gy fractions on 5 days per week for 6 weeks
• Concurrent oral TMZ at a dose of 75 mg/m2/day, 7 days per week for approximately 6 weeks
• Subjects will be randomized 1:2 to either placebo (0.9% Sodium Chloride Injection, USP, normal saline) or infusion of NanO2 (2% w/vol emulsion)
• Subjects will continuously breathe 100% oxygen for the duration of each radiation therapy treatment.
• Subjects will have a 4-week break from treatment following chemoradiation and will then re-commence single-agent TMZ per the Temodar® labeling.
• If the results from Phase 2 are promising further subjects will be recruited into Phase 3. The endpoints and objectives for Phase 3 are the same as for Phase 2.
Study population
Adult subjects (18 years and older) with recently diagnosed primary or secondary glioblastoma multiforme. In the phase 2 component of the study a maximum of 87 subjects will be enrolled at up to 20 clinical sites. Subjects will be randomized on a 1:2 basis such that 29 subjects will be randomized to standard of care + placebo and 58 randomized to standard of care + NanO2. Enrollment will be stratified according to 1) the results of methylation of the MGMT gene assayed on tumor specimen and 2) RTOG RPA.
If the results from the phase 2 component of the study are promising, recruitment into the phase 3 component will commence. At least a further 207 subjects will be randomized in a 1:2 ratio.
Dose
The dose will be 0.1 mL/kg NanO2 (2% w/vol emulsion) or 0.1mL/kg of placebo (0.9% Sodium Chloride Injection, USP, normal saline). The medical monitor shall have the power to modify or discontinue the dose of NanO2 if they determine that participant safety is at risk. The dose will reduce to either 0.075 mL/kg or 0.05 mL/kg at the discretion of the medical monitor, based on the safety and tolerability at 0.1 mL/kg and results of imaging PD biomarker studies.
Inclusion criteria
1. Histologically confirmed, newly diagnosed primary or secondary glioblastoma multiforme.
2. Treatment plan includes 60 Gy of focal radiation administered in 30 fractions, concurrently with temozolomide chemotherapy.
3. Manageable risks associated with potential radiation necrosis in the radiation field, based on size of the field and proximity to eloquent brain regions (as assessed by the investigator).
4. Aged 18 years and older.
5. Karnofsky Performance Status ≥ 70
6. Life expectancy of at least 3 months.
7. Able to undergo gadolinium-enhanced MRI (Gd-MRI) scans.
8. Baseline MRI performed within 7 days before starting study treatment while on a stable or decreasing glucocorticoid dose for at least 7 days before and during the imaging study.
9. Adequate hematologic, renal and hepatic function, as defined by:
a. Absolute neutrophil count (ANC) ≥ 1.5 x 109/L
b. Platelet count ≥ 75 x 109/L
c. Hemoglobin ≥ 10.0 g/dl
d. Serum creatinine < 1.5 x ULN
e. Total bilirubin within normal limits (≤ 2.5 x ULN if Gilbert’s syndrome)
f. Aspartate transaminase (AST) and Alanine transaminase (ALT) < 2.5 x ULN
1. Women of childbearing potential or men with child-bearing potential partners (unless vasectomized) must agree to use a highly-effective method of birth control from study entry until 4 months after completing study therapy.
2. Ability to understand and the willingness to sign a written informed consent document.
Exclusion criteria
1. Recurrent Glioblastoma
2. Prior treatment for glioblastoma apart from surgical resection.
3. Presence of multi-focal glioblastoma disease that cannot be encompassed into a radiation treatment field that would be safely treated to the prescribed radiation dose.
4. Presence of leptomeningeal disease that cannot be encompassed within a feasible and safe radiation field.
5. Intracranial bleeding, except for stable grade 1 hemorrhage or a post-operative bleed that is clearing.
6. Has not recovered from the adverse effects of surgical resection or biopsy, except for neurological deficits.
7. Subjects who have received any other investigational agent within 4 weeks before enrollment
8. Stroke or transient ischemic attack requiring hospitalization within 6 months before enrollment.
9. Myocardial infarction (MI) within 6 months before enrolment, unstable angina, New York Heart Association (NYHA) class II or greater congestive heart failure, or uncontrolled hypertension (systolic BP > 160 mmHg and/or diastolic BP > 100 mmHg).
10. Known History of Congenital long QT syndrome (12-lead EKG is not required).
11. Clinically significant chronic obstructive pulmonary disease or asthma.
12. Active major infection requiring treatment.
13. A history of other malignancies, except adequately treated non-melanoma skin cancer, curatively treated in-situ cancers or other solid tumors curatively treated with no evidence of disease for ≥ 2 years.
14. Known infection with human immunodeficiency virus or hepatitis B or C virus (testing is not required).
15. Current anticoagulant or antiplatelet therapy, except for prophylactic doses of low molecular weight heparins, low-dose aspirin, rivaroxaban (Xarelto®), apixaban (Eliquis®), or dabigatran (Pradaxa®).
16. History of allergic reactions attributed to compounds of similar chemical composition to NanO2.
17. Women who are pregnant or breast feeding.
18. Inability to comply with study procedures
19. History or evidence of any other clinically significant condition that, in the opinion of the investigator, would pose a risk to subject safety or interfere with study procedures, evaluation or completion.
20. Known hypersensitivity to any temozolomide component or to dacarbazine (DTIC).
Study assessments
Study assessments are summarized in the Study Schedule of Events. Screening procedures will be performed within 28 days of first study treatment. Safety will be assessed by weekly evaluation of adverse events, physical examinations, vital signs, pulse oximetry, and clinical laboratory tests during chemoradiation Cycle 1. Assessments are monthly during Recovery Cycle 2 and Adjuvant TMZ Cycles 3 to 8. Objective responses and progression-free survival will be assessed with contrast enhanced Gd-MRI scans using mRANO criteria, in conjunction with neurological examinations and determination of glucocorticoid doses.
MRI-based imaging biomarkers (including TOLD MRI) will be used to evaluate tumor oxygenation before and after dosing with NanO2 or placebo during the first week of therapy. Functional Assessment of Cancer Therapy – Brain (FACT-Br) Version 4 and Caregiver Quality of Life -Cancer (CQOLC) questionnaires will be used to study patient and caregiver quality of life.
Study Design
This is a randomized, prospective, placebo-controlled trial.
• All subjects will receive standard chemoradiation consisting of 30 fractions of focal brain radiation
• Total 60 Gy, given as 2 Gy fractions on 5 days per week for 6 weeks
• Concurrent oral TMZ at a dose of 75 mg/m2/day, 7 days per week for approximately 6 weeks.
• Subjects will be randomized 1:2 to either placebo (0.9% Sodium Chloride Injection, USP, normal saline) or infusion of NanO2 (2% w/vol emulsion)
• Subjects will continuously breathe 100% oxygen for the duration of each radiation therapy treatment.
• Following the 6 weeks of chemoradiation, subjects will have a 4-week break following chemoradiation treatment and will then re-commence single-agent TMZ per the Temodar® labeling.
Primary objective
1. To determine progression free survival (PFS) in newly-diagnosed glioblastoma patients after treatment with NanO2 in combination with radiation and temozolomide
Secondary objective
1. To determine overall survival after treatment with NanO2 in combination with radiation and temozolomide
2. To determine the objective response rate to study therapy using the modified Response Assessment in Neuro-oncology (mRANO) criteria
3. To determine the effect of NanO2 on the rate of pseudoprogression
4. To confirm that NanO2 re-oxygenates glioblastoma multiforme
5. To estimate the effect on the duration of functional independence as measured on the Karnofsky Performance Scale
6. To use questionnaires to study patient quality of life using FACT-Br
7. To use questionnaires to study caregiver quality of life using CQOLC
Exploratory objectives
1. To use MRI technologies to characterize the in-vivo effects of NanO2 on tumor oxygenation as a pharmacodynamic biomarker
2. To use FACT-Br and CQCOLC quality-of-life questionnaires to gain patient and caregiver perspectives for benefit/risk analysis
Subject Selection
Inclusion criteria
1. Histologically confirmed, newly diagnosed primary or secondary glioblastoma multiforme
2. Treatment plan includes 60 Gy of focal radiation administration in 30 fractions, concurrently with temozolomide chemotherapy.
3. Manageable risks associated with potential radiation necrosis in the radiation field, based on size of the field and proximity to eloquent brain regions (as assessed by the investigator).
4. Aged 18 years and older.
5. Karnofsky Performance Status ≥ 70.
6. Life expectancy of at least 3 months.
7. Able to undergo gadolinium-enhanced MRI (Gd-MRI) scans.
8. Baseline MRI performed within 28 days before starting study treatment, while on a stable or decreasing glucocorticoid dose for at least 7 days before and during the imaging study.
9. Adequate hematologic, renal and hepatic function, as defined by:
a. Absolute neutrophil count (ANC) ≥ 1.5 x 109/L
b. Platelet count ≥ 75 x 109/L
c. Hemoglobin ≥ 10.0 g/dl
d. Serum creatinine < 1.5 x ULN
e. Total bilirubin within normal limits (< 2.5 x ULN if Gilbert’s syndrome)
f. Aspartate transaminase (AST) and Alanine transaminase (ALT) < 2.5x ULN
1. Women of childbearing potential or men with child-bearing potential partners (unless vasectomized) must agree to use a highly-effective method of birth control from study entry until 4 months after completing study therapy.
2. Ability to understand and the willingness to sign a written informed consent document.
Exclusion criteria
1. Recurrent glioblastoma
2. Prior treatment for glioblastoma apart from surgical resection.
3. Presence of multifocal glioblastoma that cannot be encompassed into a radiation treatment field that would be safely treated to the prescribed radiation dose
4. Presence of leptomeningeal disease that cannot be encompassed within a feasible and safe radiation field.
5. Intracranial bleeding, except for stable grade 1 hemorrhage or a post-operative bleed that is clearing.
6. Has not recovered from the adverse effects of surgical resection or biopsy, except for neurological deficits.
7.Subjects who have received any other treatment with an investigational agent within 4 weeks before enrollment.
8. Stroke or transient ischemic attack requiring hospitalization within 6 months before enrollment.
9. Myocardial infarction within 6 months before enrollment, unstable angina, New York Heart Association class II or greater congestive heart failure, or uncontrolled hypertension (systolic BP > 160 mmHg and/or diastolic BP > 100 mmHg).
10. Known history of congenital long QT syndrome (12-lead EKG is not required).
11. Clinically-significant chronic obstructive pulmonary disease or asthma.
12. Active major infection requiring treatment.
13. A history of other malignancies, except adequately treated non-melanoma skin cancer, curatively treated in-situ cancer or other solid tumors curatively treated with no evidence of disease for ≥ 2 years.
14. Known infection with human immunodeficiency virus or hepatitis B or C virus (testing is not required).
15. Current anticoagulant or antiplatelet therapy, except for prophylactic doses of low molecular weight heparins, low-dose aspirin, rivaroxaban (Xarelto®), apixaban (Eliquis®), or dabigatran (Pradaxa®).
16. History of allergic reactions attributed to compounds of similar chemical composition to NanO2.
17. Women who are pregnant or breast feeding.
18. Inability to comply with study procedures.
19. History or evidence of any other clinically-significant condition that, in the opinion of the investigator, would pose a risk to subject safety or interfere with study procedures, evaluation or completion.
20. Known hypersensitivity to any temozolomide component or to dacarbazine (DTIC).
Registration Procedures
An Eligibility Screening Worksheet with a Registration Form must be completed by the participating site and forwarded to the Sponsor Once Sponsor has approved the subject, site staff will enter the subject into the electronic data capture (EDC) system. The EDC will be used to collect all protocol defined patient data and to randomize the subject to treatment (see Section 6.5 below for details).
Issues that would cause treatment delays should be discussed with the site Principal Investigator. If a subject does not receive protocol therapy following registration, the subject will be considered a screen failure and replaced. Screen failures will be replaced and will not be included in the any analysis.
Registration documents
The following registration documents must be completed by the Principal Investigator or Designee at the participating site and forwarded to Sponsor prior to enrolling each subject.
• Signed Signature page(s) of the Subject Consent Form
• Completed Eligibility Screening Worksheet
• Completed Registration Form
Treatment Plan
After recovery from surgical resection (or biopsy in non-resectable patients), all subjects will receive standard front-line therapy for GBM involving focal radiation (60 Gy in 30 fractions over 6 weeks) with concurrent and adjuvant TMZ chemotherapy [2]. During the 6 weeks of concurrent chemoradiation, NanO2/placebo doses will be administered by IV push with each fraction of radiation. Following completion of 6 cycles of adjuvant TMZ, subjects will then be monitored with serial MRI scans (as per standard of care) until progressive disease.
NanO2/Placebo
NanO2/placebo will be administered at 0.1mL/kg by IV push over a period of no more than 10 minutes. Additionally, delivery should be completed within 15-60 minutes, but no more than 90 minutes, prior to each fraction of radiation. Every effort should be made to infuse within the same time parameters of the first dose for consistency of data. No premedication is needed. Each dose will be prepared on the day of administration. Dose preparation and handling are described in detail in the Pharmacy Manual. NanO2/placebo may be administered via an IV catheter or peripheral IV cannula placed in a peripheral vein.
Oxygen
All subjects will continuously breathe 100% oxygen from the start of each NanO2 or placebo infusion through completion of the radiation fraction scheduled for the same day. 100% oxygen will be provided by the treating institution in portable cylinder or from a wall outlet supply. On day 1 of the chemoradiation treatment, subjects will have baseline TOLD MRI scans pre and post infusion and will continue to breathe 100% oxygen from completion of the radiation fraction until completion of the TOLD MRI scans. Note that for the baseline TOLD MRI scan and follow-up TOLD MRI scan, the subjects should breathe oxygen during the scan. If a tank of gas is used, the tank must be MRI compatible. [3].
Radiation therapy
All subjects will receive standard focal brain radiation for a total dose of 60 Gy in 30 fractions (2 Gy fractions daily, 5 days per week over 6 weeks) using a linear accelerator. Subjects will be in a supine position and immobilized in a thermoplastic cast for planning and treatment. The radiotherapy planning CT scan will be fused with post-operative MRI images and contouring will also be guided by pre-operative MRI imaging. The Gross Tumor Volume (GTV) is defined as the tumor cavity plus any residual contrast-enhancing tumor. The GTV will be expanded isotropically by 1.5 cm to form the Clinical Target Volume (CTV), which will be curtailed along natural barriers to subclinical tumor spread (such as the falx). A further isotropic expansion of 3 mm will be performed to reach the Planning Tumor Volume (PTV). Conformal planning will involve a minimum of three shaped beams to produce PTV coverage by the 95% isodose with dose homogeneity of 95-105%. Maximum point doses within optic structures and the brainstem will be limited to 55 Gy. The primary field is targeted to the T2/Flair images + 1.5 cm CTV margin + 3 mm PTV margin to create the first XRT field receiving 46 Gy. Then there is a sequential boost of 14 Gy given to the T1+C images + 1.5 cm CTV + 3 mm PTV. Alternatively, a simultaneous integrated boost (SIB) technique to the same volumes and doses, respectively, is permitted. Intensity-Modulated Radiation Therapy (IMRT) is desirable, but 3D-CRT is acceptable provided the same dose coverage and organ tolerances are met. Radiation fractions will be delivered within 15-60 minutes after completion of each NanO2/placebo infusion (under 90 minutes is acceptable).
Concurrent Temozolomide chemotherapy
Concurrent with the radiation therapy, subjects will receive oral TMZ at a dose of 75 mg/m2 once daily on 7 days per week. Doses will be taken in the morning, before radiation fractions, on an empty stomach (at least 45 minutes before breakfast). The chemotherapy will continue for the duration of the radiation or for 42 days, whichever is shorter. Following completion of chemoradiation, subjects will have a 4-week break from treatment. Subsequently, TMZ will commence on an intermittent schedule, with doses administered once daily on days 1-5 of a 28-day cycle. The TMZ dose will be 150 mg/m2/day in cycle 1 and 200 mg/m2/day in subsequent cycles, and a total of 6 cycles will be administered. Doses will be rounded to the nearest 5 mg. The next cycle of TMZ will not commence until ANC≥ 1.5 x 109/L and platelets ≥ 100x109/L. See Section 6.6 for recommended antiemetic pre-medication and Section7 for recommended dose reductions for toxicity.
Method of assigning subjects to treatment groups
Randomization and blinding
A computer-generated randomization algorithm will be used to randomize the subjects in a 1:2 ratio to the placebo or NanO2 treatment groups. Randomization will be stratified by Methylation status (positive versus negative) of the MGMT gene determined by the analysis of the archived tumor specimens from all subjects and by the ROTG-RPA results (class III/IV vs class V).
Once a subject has been accepted into the study via the registration process and allocated a study number, they will be randomized to treatment using the randomization module in the EDC system. The EDC system will advise the randomization number. This is a double blinded study.
Patients, treating physicians, assessing physicians, Investigators. study personnel and the medical monitor will be blinded. Only the pharmacist preparing the study drug or placebo will be unblinded.
The unblinded pharmacist will be provided the treatment group that each patient is assigned to , and will dispense the blinded treatment to the study personnel who will administer the drug to the subject. Refer to Section 10.9 if unblinding is necessary.
Concomitant medication and supportive care guidelines
In general, all supportive measures consistent with optimal patient care will be given throughout the trial and reported in the CRF. However, the following restrictions apply:
• If an indication for therapeutic anticoagulation develops on study, only low molecular weight heparins (e.g., enoxaparin) should be used.
• If a patient uses Tumor Treating Fields (TTF), such as Optune®, as a treatment modality, the use of this device must be recorded in the CRF as well as the patient adherence to this device.
Prophylactic antiemetic pre-medication with a selective 5-HT3 receptor antagonist (e.g., ondansetron 8 mg orally) is recommended prior to each TMZ dose from the beginning of concurrent chemoradiation and during adjuvant treatment. Constipation is likely to result from prolonged administration of a 5-HT3 receptor antagonist and appropriate therapy should be commenced as required. During concurrent chemoradiation (when the daily TMZ is relatively low), investigators may choose to substitute metoclopramide or no anti-emetic treatment in place of a 5-HT3 receptor antagonist once good tolerance of chemoradiation is apparent.
Subjects developing neutropenic fever or infections will be treated with broad-spectrum antibiotics according to usual practice at the treating institution. Transfusion therapy for thrombocytopenia and anemia will be performed as per institutional guidelines. Symptomatic radiation necrosis will be managed using glucocorticoid therapy and other measures as per investigator discretion. Recommended dose delays and dose reductions for hematological and non-hematological toxicities are described in Section 7. G-CSF or other hemopoietic growth factors should not be given routinely but may be used to treat an episode of neutropenic fever or severe prolonged neutropenia at the investigator’s discretion.
All subjects will be given standard prophylactic medication for pneumocystis pneumonia (PCP) during Cycle 1 as described in the temozolomide labeling unless there is a medical contraindication. Prophylactic PCP medication should be continued in patients who experience lymphopenia until resolution to Grade 1 or less. The medication regimens for PCP prophylaxis may be altered, including decreasing or discontinuing administration at the treating physician’s discretion.
Adjuvant temozolomide treatment
Temozolomide treatment is to be administered once daily on Days 1 to 5 of each 28-day cycle for 6 cycles, Cycle 3 to Cycle 8, per Standard of Care.
Dosage in Cycle 3 is 150 mg/m2 once daily for 5 days. At the start of Cycle 4, the dose can be escalated to 200 mg/m2. The dosage in Cycles 4 to 8 may be increased to 200 mg/m2 per day if the following conditions are met before starting cycle 4. If the dose was not escalated at the onset of Cycle 4, do not increase the dose for Cycles 5 to 8.
• Nonhematologic toxicity is Grade 2 or less (except for alopecia, nausea, and vomiting)
• ANC is greater than or equal to 1.5 × 10 /L, and
• Platelet count is greater than or equal to 100 × 10 /L.
Duration of adjuvant temozolomide therapy
In the absence of treatment delays due to adverse events, TMZ treatment may continue as scheduled until one of the following criteria applies:
• Disease progression
• Intercurrent illness that prevents further administration of treatment
• Unacceptable adverse event(s)
• Subject decides to withdraw from the study
• Changes in the subject's condition that render continuing treatment unacceptable, per the investigator’s judgement.
Duration of follow-up
All subjects undergoing at least one dose of study therapy and have not withdrawn consent to participate in the study will continue in the follow-up phase for disease progression and survival assessment. Onsite follow up visits at 3 month intervals will start 3 months after a subject discontinues study therapy, or the last adjuvant TMZ treatment. The onsite visits will continue until disease progression is identified. Remote follow up visits will start after disease progression and will continue at 3 month intervals for a total of 3 years after the last administration of study drug. Remote follow up visits will be made every 6 months for an additional 2 years. Therefore, all subjects will be followed for 5 years after the last administration of study drug.
End of study
All subjects exiting the study early will undergo an end of study visit to ensure that they safely exit the study. Assessments scheduled for Cycles 3 to 8 do not need to be repeated at the end of study visit if they were performed within 14 days of the end of study visit.
Dosing Delays and Dose Modifications
NanO2 dose modifications
The medical monitor shall have the authority to modify or discontinue the dose of NanO2 if they determine that participant safety is at risk. The dose will be reduced to either 0.075 mL/kg or 0.05 mL/kg at the discretion of the medical monitor , based on the safety and tolerability at 0.1 mL/kg and results of imaging PD biomarker studies. Subjects receiving reduced doses will remain on study. Investigators will report all adverse events related to NanO2 to the medical monitor as described in Section 10. DLTs and related stopping criteria are described in Section 9.
TMZ dose modifications
Dose reductions in TMZ for toxicity (i.e., adverse events considered related to TMZ) will be implemented per the Temodar labeling. During the concomitant phase, dose reductions are not recommended. However, dose interruptions or discontinuation may occur based on toxicity.
If the toxicity for Cycle 3 is Grade ≤ 2 (except for alopecia, nausea, and vomiting), absolute neutrophil count (ANC) ≥ 1.5 x 109/L and the platelet count ≥ 100 x 109/L. The dose remains at 200 mg/m2 for the remaining cycles, except if toxicity occurs. If the dose was not escalated in Cycle 4, it should not be escalated in subsequent cycles. Dose reductions or discontinuations during the maintenance phase should be applied according to Table 1-3.
Table 1. Comparison of Safety Profile of NanO2 Versus Fluosol as a Radiosensitizer
| Agent | w/vol % FC | "Dose/kg (gFC/kg) | Indication | "Frequency Dosing (Total doses)" | Acute complications |
|---|---|---|---|---|---|
| NanO2 [18] | 2 | 0.1 mL (0.002) | GBM | 5/week, 30 doses | The only definite TEAE was a brief episode of dizziness that occurred in 1 subject. Two subjects had radiation necrosis that was deemed probably related to NanO2. The attached Adverse Events Report lists all of the potential adverse events in the trial and shows that administration of NanO2 is safe in association with chemoradiation. |
| Fluosol [19] | 20 | 8.0 mL (1.6) | Head and Neck | 1/week, 5 doses | 4 of 15 subjects experienced flushing, warmth or chest pressure with first dose. 1 subject had fever to 38.5o C. Increased liver enzymes, alk phos, SGOT and SGPT in 8/15 subjects. Alk phos remained elevated for about 3 months. |
| Fluosol [20] | 20 | 8.0 mL (1.6) | GBM | 1/week 7 doses | 2/3rds of subjects had allergic reaction, 11 of 18 subjects had elevations in alk phos, SGOT and SGPT. Liver function abnormalities persisted through treatment. Most resolved by 3 months |
| Fluosol [21] | 20 | 10.33 – 15.50 mL (2.1–3.1)* | Recurrent GBM | 1/week 6 doses | 98 subjects were enrolled. Alterations of vital signs occurred in sixty-eight subjects (69.4%) within 5 or 10 minutes of Fluosol test dose or infusion. At least one non-lethal Fluosol-related adverse event occurred in 32% of the 98 subjects evaluable for safety (76 occurrences). Most frequently reported were Fluosol-related nausea/ vomiting (16 occurrences), flushing, dyspnea, and chest tightness/chest pain (7 occurrences each) and back pain (4 episodes). Six events were grade 3 |
*The dose reported in this study was converted from the cumulative dose 400 – 600 mL per M2 assuming 60 kg body weight corresponds to 1.55 M2.
Table 2. TMZ dose levels for toxicity modifications during adjuvant treatment
| Dose Level | TMZ Dose |
|---|---|
| 1 | 200 mg/m2 days 1-5, Dose during Cycles 2-6 in absence of toxicity |
| 0 | 150 mg/m2 days 1-5, Dose during Cycle 1 |
| 1 | 100 mg/m2, Reduction for prior toxicity |
Table 3. Dose Reductions
| Toxicity | Reduce TMZ by 1 Dose Level * | Discontinue TMZ |
|---|---|---|
| Absolute Neutrophil Count | Less than 1.0 x 109/L | See footnote * |
| Platelet Count | Less than 50 x 109/L | See footnote * |
| CTC Nonhematological Toxicity (except for alopecia, nausea, vomiting) | CTC Grade 3 | CTC Grade 4* |
During treatment, a complete blood count will be obtained on Cycle 3 Day 22 (21 days after the first adjuvant dose of TMZ) or within 48 hours of that day, and weekly until the ANC is above 1.5 x 109/L. The next cycle of adjuvant TMZ should not be started until the ANC and platelet count exceed those levels.
Study Assessments
Refer to the Appendix C: Schedule of Events for a summary of study procedures required at each visit. Additional procedures deemed necessary as part of standard care may be performed at the investigator’s discretion.
Screening and informed consent
Study-specific screening procedures may only commence once a subject has provided written informed consent. However, procedures that are part of routine care may be used to determine eligibility if they comply with protocol requirements.
Specific procedures
Height and weight
Measured in light-weight clothing, without shoes.
Vital signs and pulse oximetry
Vital signs include temperature and resting (sitting for at least 5 minutes) pulse rate, respiration rate, blood pressure, and pulse oximetry.
Physical examination
Physical examinations will be performed by the PI or medically-qualified designee.
The complete physical examination will include evaluation of skin, HEENT, lymph nodes, heart, chest, abdomen and extremities, and a neurological examination (to include assessment of speech, cranial nerves, motor power, deep tendon reflexes, sensation, coordination, and gait).
The abbreviated physical examination will include assessment of skin, oral cavity, heart, chest, abdomen and extremities, and additional focused assessments suggested by the presence of specific symptoms.
Clinical laboratory assessments
Laboratory tests will be performed by the local clinical laboratory.
Serum chemistry
Blood chemistry will include sodium, potassium, BUN, creatinine, calcium, glucose, total bilirubin, alkaline phosphatase, AST, ALT, albumin and total protein. Creatinine clearance will be calculated using the Cockcroft-Gault formula for subjects with serum creatinine outside the normal range.
Cockcroft-Gault formula
CG Creatinine clearance = [(140-age) x weight x 0.85 if female] / (72 x Scr), adjusted for BSA by 1.73m2 / BSA Where: Scr is serum Creatinine
Hematology
Hematology will include hemoglobin, hematocrit, red cell count, MCV, MCH, MCHC, white cell count with differential count (neutrophils, basophils, eosinophils, lymphocytes and monocytes) and platelet count.
Pregnancy test
A serum pregnancy test will be performed in women of childbearing potential.
Predictive biomarker studies on archived tumor
MGMT and IDH1 will be evaluated at the sites’ local laboratory prior to study treatment. Up to ten archived formalin-fixed paraffin-embedded GBM tissue from the initial presentation will be prepared and shipped to the sponsor as described in the Laboratory Manual. The sponsor will store the slides which may be evaluated for biomarkers.
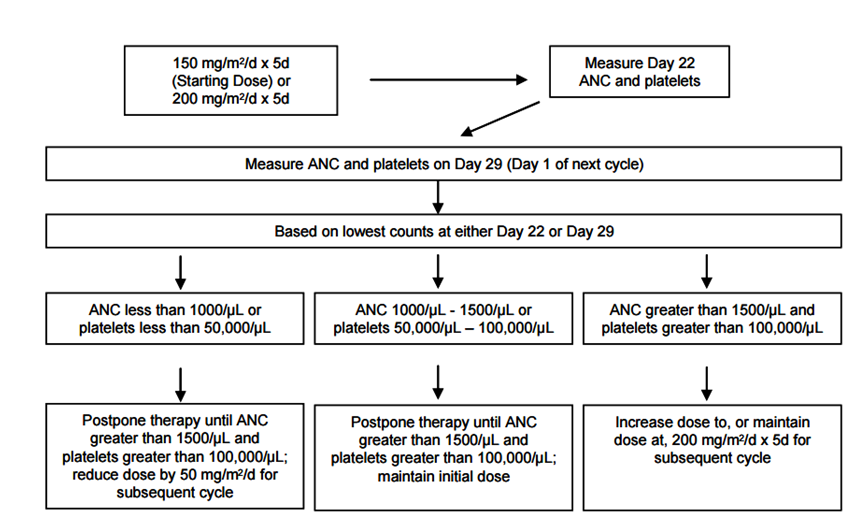
Figure 1:Dosing Modification Table from Temodar Package Insert
Quality of Life Recorded Outcomes
Quality of life questionnaires will be used to gain patient and caregiver perspectives. Questionnaires will be collected prior to treatment on the same day as Dose 1 and Dose 16 of Cycle 1-chemoradiation treatment, Day 28 of Cycle 2-Recovery Period, Day 1 of Cycle 6 and Day 28 of Cycle 8 of the maintenance phase, through long-term study follow up, and at the end of study visit.
• Functional Assessment of Cancer Therapy – Brain (FACT-Br) Version 4 [28]
• Caregiver Quality of Life – Cancer (CQOLC) [29]
Magnetic resonance imaging
MRI scans are to be obtained as per standard of care (except for the addition of TOLD MRI scans). A gadolinium-enhanced MRI (Gd-MRI) scan of the brain will be performed during screening within 7 days of the first radiation therapy treatment to be used as the baseline scan. Subsequently, scans will be repeated 3-4 weeks after completion of chemoradiation and every 8-weeks during adjuvant TMZ. During follow-up, MRI scans will be repeated every 3 months. After 3 years post therapy completion, scans will be repeated every 6 months during follow-up. MRIs will be performed as described above until disease progression.
To summarize, all MRI Scans are to be scheduled on days:
• Screening and/or Post-operative
• Cycle 2 Day 28 of Recovery Period
• Cycle 4 Day 28 of Adjuvant TMZ
• Cycle 6 Day 28 of Adjuvant TMZ
• Cycle 8 Day 28 of Adjuvant TMZ
• Follow-up every 3 months (+/- 7 days) for 3 years
• Long-term follow-up at every 4-6 months post 3 years
Scans will be acquired with slice thickness of 5 mm or less and will include T1- and T2-weighted images, axial flair, post-contrast gradient echo scans in all three planes and diffusion- and susceptibility-weighted images. Subsequent scans will be performed in the same manner as at baseline and preferably on the same scanner. Additional information can be found in the Image Acquisition Protocol.
Tumor response will be assessed using the modified Response Assessment in Neuro-oncology (mRANO) criteria in Section 12.2.Any subject with symptoms suggestive of disease progression should be promptly evaluated with a repeat MRI scan. All scans will be uploaded to, a secure, cloud-based medical image management platform by the study sites. Additional information can be found in the Imaging Charter.
Assessment of the mRANO-criteria response by the treating oncologist and site radiologist will be used to make all study treatment decisions.
Imaging PD biomarkers
MRI scans incorporating sequences that provide informative information regarding the oxygenation of tumor tissue and surrounding normal brain (TOLD MRI) will be performed immediately before and within 180 minutes after completing the NanO2/placebo infusion. ideally on day 1 of chemoradiation, but it is permitted that this be obtained on days 1-5, i.e., during the first week of treatment. It is preferable for the scans to be completed within 120 minutes whenever possible. Both TOLD MRI scans must be completed on the same day.
No gadolinium is to be administered on days when TOLD MRI scans are performed
unless a gadolinium enhanced scan is deemed indicated by the investigator, and then gadolinium can only be administered after completion of all TOLD scans.
Study sites will directly upload images to a secure, cloud-based medical image management platform by the study sites. Note that no formal read of the TOLD MRI is to be performed by the study sites. as this will be performed by the imaging core lab.
Assessments performed by study visit
Screening Day 0 (Window: -28 to -1)
Before any study-specific procedures are performed, potential study subjects must complete the informed consent process, including signing the Subject Information and Consent Form approved by the IRB. The following assessments, unless otherwise stated, will then be performed within 28 days before the first dose of study treatment:
• Medical history, including demographics, detailed neuro-oncology history including previous brain-tumor treatments, medication history and past relevant medical and surgical history
• Height and weight
• Vital signs and pulse oximetry
• KPS
• Complete physical examination
• Review of concomitant medications
• Clinical laboratory tests, including serum chemistry, complete blood count and a serum pregnancy test for women of child-bearing potential. Pregnancy test to be performed within 2 days of Dose 1.
• Predictive biomarker studies on archived GBM tissue. The study used may be from a time prior to the 28-day screening period. Note the biomarker study is not an eligibility criterion but subjects will be stratified based on the results.
• Baseline Gd-MRI scan (only performed if radiation is scheduled to commence later than 7 days after previously obtained scan.).
• Determination of eligibility
• Randomization to treatment group within 5 days of scheduled study chemoradiation
Cycle 1 – Chemoradiation Treatment
Subjects will receive 6 weeks of treatment 5 days a week with NanO2 or placebo combined with standard chemoradiation therapy for a total of 30 doses.
Subjects will continuously breathe 100% oxygen for the duration of each NanO2 or placebo infusion and until the end of each radiation therapy treatment. Oxygen therapy will continue during TOLD imaging on Cycle 1 Dose 1 as described in Section 6.2.
Dose 1
Before NanO2/placebo administration
• Weight
• Vital signs and pulse oximetry
• Abbreviated physical examination
• Review of concomitant medications
• Serum chemistry and complete blood count. Results must be reviewed by the Investigator prior to dosing (can be performed ≤ 1 calendar day prior to the visit )
• FACT-Br and CQOLC questionnaires
• Baseline imaging PD biomarker scan (TOLD MRI) (can be performed on one day with pre and post imaging on day 1 or another day during the first week)
• Adverse event collection prior to Dose 1 is limited to events experienced by the subject after informed consent, and related to procedures required by the protocol which the subject would not have otherwise underwent if not participating in the study.
During and after NanO2/placebo and radiation therapy administration
• Vital signs and pulse oximetry performed post NanO2/placebo infusion
• Repeat imaging PD biomarker scan (TOLD MRI) (can be performed on day 1 or another day of the first week, but baseline and follow up PD biomarker scans must be performed on the same day)
• Adverse event collection after initiation of Dose 1
Doses 2-5
Before NanO2/placebo administration
• Vital signs and pulse oximetry
• Adverse event collection
During and after NanO2/placebo and radiation therapy administration
• Vital signs and pulse oximetry
• Adverse event collection
Doses 6, 11, 16, 21, 26 and 30
Before NanO2/placebo administration
• Weight
• Vital signs and pulse oximetry
• Abbreviated physical examination
• Review of concomitant medications
• CBC every week. Serum chemistry lab results at dose 16 and 30 must be reviewed by a study team Investigator prior to dosing (can be performed ≤ 1 calendar day prior to the visit)
• FACT-Br and CQOLC questionnaires (Dose 16 only)
• Adverse event collection
During and after NanO2/placebo and radiation therapy administration
Vital signs and pulse oximetry will be performed after commencing NanO2/placebo
Adverse event collection
Doses 7 – 10, 12 – 15, 17 – 20, 22 – 25 and 27 – 29
• NanO2/placebo and radiation therapy administration
• Adverse event collection
Cycle 2 – RECOVERY PERIOD – Post-chemoradiation
Subjects will undergo a 28 day recovery period post chemoradiation where no cancer related treatments will be administered. Subjects will be followed periodically during this period. Standard practices suggest that the first post-chemoradiation visit will be the first day in the week following the last chemoradiation dose which is nominally on day 3. Subjects will be seen in clinic on Day 28 of the recovery period.
Day 3, 11, 18
• Telephone follow up
• Review of concomitant medications
• Adverse event collection
Cycle 2 Day 28: Prior to Cycle 3 Adjuvant Temozolomide
Weight
• Vital signs
• KPS
• Complete physical examination
• Review of concomitant medications
• Serum chemistry and complete blood count performed between days 22 and 28 per standard care
• Adverse event collection
• FACT-Br and CQOLC questionnaires
• Gd-MRI scan
Adjuvant temozolomide cycles
Adjuvant phase consists of 6 cycles as described in Section 6.7.
Day 1 to 5 of Cycle 3-8
• Adjuvant TMZ treatment
Day 28 of cycles 3-7: prior to next adjuvant cycle
• Vital signs
• Abbreviated physical examination
• Serum chemistry and complete blood count performed between days 22 and 28 per standard care
• Adverse event collection
• FACT-Br and CQOLC questionnaires only on Day 28 of Cycle 5
• GD-MRI only on Day 28 Cycles 4 & 6
Day 28 of cycle 8
• Vital signs
• KPS
• Abbreviated physical examination
• Serum chemistry and complete blood count performed between days 22 and 28 per standard care
• Review of concomitant medications
• Adverse event collection
• FACT-Br and CQOLC questionnaires
• GD-MRI
Onsite Follow-up visits every 3 months ± 14 days until disease progression
• Weight
• Vital signs
• KPS
• Complete physical examination
• Review of concomitant medications
• Adverse event collection
• Serum chemistry and complete blood count only per standard care
• FACT-Br and CQOLC questionnaires
• Gd-MRI scan
Remote Follow up visits every 3 months ± 14 days for 3 years after disease progression
Remote visits can be made by telephone call, clinic visit, through another physician or via registry search.
• KPS
• Survival Assessment
Remote Follow up visits every 6 months ± 30 days for an additional 2 years
Remote visits can be made by telephone call, clinic visit, through another physician or via registry search.
• KPS
• Survival Assessment
End-of-study visit
An end of study visit is to be performed for subjects ending study participation prior to the identification of disease progression with the exception that KPS assessment is to be performed for subjects ending study participation after progression of disease has been identified.
• Weight
• Vital signs
• KPS
• Complete physical examination
• Review of concomitant medications
• Serum chemistry and complete blood count only per standard care
• Adverse event collection
• FACT-Br and CQOLC questionnaires
Dose Limiting Toxicities
Dose Limiting Toxicity (DLT)
A DLT is defined as any of the following events occurring within 4 hours of initiation of a NanO2 infusion and deemed by the investigator to be possibly, probably or definitely related to NanO2 that result in modification of the NanO2 dose:
• Adverse events of NCI CTCAE Grade 3 or 4 severity;
• Serious adverse events as defined in Section 10;
• Adverse events which result in an interruption or premature discontinuation of temozolomide or planned radiation.
Dose reductions and stopping criteria
In the event of a DLT, no further administration of NanO2 will be given to the subject until the DLT resolves and the medical monitor has determined it is acceptable to continue dosing in the subject who had the DLT. Where medically appropriate, standard of care radiation and temozolomide will be continued during any interruption of NanO2. The medical monitor may recommend continuing dosing at a dose reduction of either 0.075 mL/kg or 0.05 mL/kg in response to the DLT. If the medical monitor does not recommend continuing dosing, the subject who had the DLT will be discontinued from study treatment and returned to his/her physician for standard care of treatment and will continue follow up according to the protocol.
Adverse Events
An adverse event (AE) is defined as any untoward medical occurrence in a clinical trial subject. The event does not necessarily have a causal relationship with study treatment. Worsening of a preexisting medical condition is also defined as an AE. Progression of the recurrent glioblastoma should not be captured as an adverse event. If a new primary malignancy appears, it will be considered an adverse event.
The investigator is responsible for reviewing laboratory test results and determining whether an abnormal value in an individual study subject represents a clinically significant change from the subject’s baseline values. In general, abnormal laboratory findings without clinical significance (based on the investigator’s judgment) should not be recorded as AEs. However, laboratory value changes that require treatment or adjustment in current therapy are considered AEs. Where applicable, clinical sequelae (not the laboratory abnormality) should be recorded as the AE.
Reporting procedures for adverse events
The investigator is responsible for ensuring that all AEs observed by the investigator or reported by the subject that occur after signing of informed consent through 30 days after the last administration of study treatment are reported using the applicable CRF. Adverse events will not be collected for screen failures, or subjects in long-term follow-up for survival.
The investigator must assign the following attributes to an AE:
• Diagnosis or syndrome(s), report signs and/or symptoms
• Dates of onset and resolution
• Severity
• Assessment of relatedness to study drug
• Action taken
A CTCAE Grade 3 or 4 acute or delayed adverse event which also meets the definition of an SAE will be filed immediately in the source document by the clinical nurse or site investigator. The event will be communicated by the site investigator in the form of an Investigator Safety Letter via email to the Sponsor and medical monitor within 7 days of the event being reported by the study staff. Additionally, an adverse event report form will be filed.
Any adverse event which is possibly, probably or definitely related to NanO2 and requires dose modification, delay or stoppage shall be reported to the Sponsor and medical monitor in the same timeframe as an SAE described in Section 10.6.
Adverse event severity
The descriptions and severity grading scales found in CTCAE version 5.0 will be utilized for AE reporting. All appropriate treatment areas should have access to a copy of the CTCAE. Grade refers to the severity of the AE. The CTCAE displays Grades 1 through 5 with unique clinical descriptions of severity for each AE based on this general guideline:
Relationship of adverse event to study treatment
Investigators will review each AE and attribute the causality of the event to the study drug according to the following categories:
Definitely: clear evidence exists that the event was caused by the study treatment; a strong temporal relationship exists, and an alternative cause is unlikely.
Probably: reasonable probability exists that the event was caused by the study treatment; the event has a strong temporal relationship to the study procedure(s) and follows a known pattern of response; an alternative cause seems unlikely.
Possibly: reasonable possibility exists that the event may have been caused by the study treatment; the event has a temporal relationship to the study procedure(s) and follows a known pattern of response, but a potential alternative cause may be present.
Unlikely: evidence exists that the event was caused by other factors; although the relationship of the event to the study treatment cannot be completely ruled out, is remote.
Unrelated: the cause of the event is known, and the event is in no way related to any aspect of the study treatment.
For determination of causality, unlikely and unrelated will be considered not related.
Follow-up of an AE
Medically-significant AEs considered related to study drug by the investigator, or the sponsor, will be followed until resolved or considered stable. The investigator’s clinical judgment will be used to determine whether a subject should be removed from treatment due to an adverse event.
Serious adverse events
Definition of a serious adverse event (SAE)
An adverse event is considered serious if it results in ANY of the following outcomes:
1. Death
2. A life-threatening adverse event
3. An adverse event that results in inpatient hospitalization or prolongation of existing hospitalization for ≥ 24 hours
4. A persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions
5.A congenital anomaly/birth defect
6. Important medical events that may not result in death, be life threatening or require hospitalization, but may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed in this definition
Reporting procedures for SAEs
The investigator at each participating site is responsible for ensuring that all SAEs observed by the investigator, study staff or reported by the subject that occur after signing of informed consent through 30 days after the last administration of study NanO2 treatment; and SAEs that occur more than 30 days after the last administration of study treatment attributed to be possibly, probably or definitely to NanO2 treatment are recorded in the subject’s medical record and are reported to the Sponsor and medical monitor within one business day of first knowledge or discovery of the event. The exception to the 30-day SAE follow up is for radiation necrosis, which will be evaluated as an AE or SAE at any time it occurs.
This initial notification will be followed by a complete report as soon as possible but in no case later than 5 calendar days of the initial report.
Sponsor will report SAEs and suspected unexpected serious adverse reactions (SUSARs) to regulatory authorities, investigators and IRBs in compliance with all reporting requirements according to local regulations and good clinical practice. Reporting of SAEs and SUSARs will be done in accordance with the timeframes set out in 21 CFR 312.32. In the case of unexpected fatal or life-threatening suspected adverse reactions, the Sponsor will notify the FDA as soon as possible but in no case later than 7 calendar days after the initial receipt of the information.
The investigator at each participating site will notify their appropriate IRBs of SAEs occurring at their site and other AE reports received from the sponsor and NuvOx, in accordance with local procedures and statutes.
If a greater than 20% rate of SAEs is observed, the DSMB will meet and determine if the trial should be modified or discontinued.
Routine reporting
All AEs must be reported in routine study data submissions. Clinical sites must report the death of subject to NuvOx until all subjects complete their participation in the study.
Data safety and monitoring plan
All moderate and severe safety signals that are unanticipated and probably or definitely related to the investigational product will be discussed with the DSMB and the Sponsor prior to enrolling additional subjects. If the study is halted due to concerns regarding subject safety, the DSMB shall have the power to modify or discontinue the dose of NanO2 if it is determined that subject safety is at risk.
Data safety and monitoring board
A Data Safety and Monitoring Board (DSMB) will be selected. The DSMB will consist of 4 members. 3 members will constitute a quorum. Membership consists of persons completely independent of the investigators and who have no financial, scientific, or other conflict of interest with the trial. All members will sign a DSMB charter signifying their understanding of all DSMB responsibilities. The DSMB will create a charter at their first meeting. The meeting schedule will be dependent upon enrollment milestones as defined in the DSMB Charter. The Sponsor shall appoint a blinded medical monitor.
Emergency unblinding
To maintain the overall quality and legitimacy of the clinical trial, randomization code breaks should only occur in circumstances when knowledge of the actual treatment is crucial for further management of the subject. An emergency unblinding procedure is built into the randomization system of the eCRF. The Investigator must confer with the medical monitor to determine if unblinding is deemed to be necessary, the Investigator will have administrative access to request unblinding for a study subject within the randomization system of the eCRF. When the Investigator unblinds the treatment for the subject, the system will record the date and time when the information was unblinded. The Investigator and medical monitor will document the reason for unblinding.
The Investigator is encouraged to maintain the blind as far as possible. The allocation must NOT be disclosed to the subject and/or other study personnel including other site personnel, monitors, corporate sponsor or project office staff; nor should there be any written or verbal disclosure of the code in any corresponding subject documents.
Pharmaceutical Information
Description of the investigational product
The Product Description of NanO2, storage conditions and stability data are presented in the Investigator’s Brochure. Clinical supplies for this study have been manufactured under aseptic conditions at NuvOx Pharma in Tucson, Arizona, USA. Solution preparation prior to administration is described in the Pharmacy Manual.
The placebo is 0.9% Sodium Chloride Injection, USP, normal saline, prepared by pharmacy staff according to instructions in the Pharmacy Manual. Immediately prior to injection, the placebo will be aseptically compounded and placed directly into the same dispensing syringes as those for dispensing used for the injection of NanO2. The placebo will not be stored longer than required for immediate administration.
Administration
NanO2/placebo will be administered at 0.1mL/kg by IV push over a period of no more than 10 minutes. Additionally, delivery should be completed within 15-60 minutes, but no more than 90 minutes prior to each fraction of radiation (total of 30 doses). Every effort should be made to infuse within the same time parameters of the first dose for consistency of data. No premedication is needed. Each dose will be prepared on the day of administration. Dose preparation and handling are described in detail in the Pharmacy Manual. NanO2/placebo may be administered via an IV catheter or peripheral IV cannula placed.
Dose modifications are described in Section 7.
The syringes containing NanO2 or placebo will be appropriately labeled for investigational use only and provided to the clinical team in a blinded manner. Subject body weight will be reported in kg, and the weight will be rounded to the nearest whole number. The weight will be taken on Day 1; this weight will be checked on a weekly basis. If the body weight changes by ≥ +/- 5%, then the dose will be recalculated.
Availability, ordering and accountability
NanO2/placebo will be made available by the Sponsor to study sites after site initiation. Investigational product will be ordered by and sent to the responsible clinical trial pharmacist, who will review and document the amount and condition of the medication and acknowledge receipt to the Sponsor. At the end of the study, any remaining used or unused investigational product will be destroyed according to institutional procedures.
Accountability records will contain the following information:
• Quantities of NanO2 and placebo received and dates of arrival
• Subject identification numbers
• Dose preparation records
• Dates of dose preparation and dispensing
• Amounts of any doses returned to pharmacy
• Disposition of unused doses
Temozolomide
TMZ is a commercially available drug considered standard of care for treating glioblastomas, and will be obtained via the treating institution’s standard purchasing channels and administered according to labeled prescribing procedures.
Measurement of Effect
Assessment of antitumor effect
For the purposes of response assessment, subjects should be evaluated by Gd-MRI scanning at baseline,3-4 weeks after completion of chemoradiation and every 8-weeks during adjuvant TMZ. During follow-up, MRI scans will be repeated every 3 months until disease progression is identified. In addition, confirmatory scans to rule out pseudo progression should be obtained not less than 4 weeks following initial documentation of disease progression .
mRANO criteria
Response and progression will be evaluated in this study using the modified Response Assessment in Neuro-Oncology (mRANO) criteria, which provides standardized response and progression criteria for use in clinical trials of treatment for newly diagnosed and recurrent glioblastoma [34]. The mRANO criteria incorporates the higher-than-normal incidence of treatment-related increase in contrast enhancement (so-called “pseudoprogression,” PsP) or decrease in contrast enhancement (so-called “pseudoresponse,” PsR). Both progression and response require a confirmation equal to or greater than 4 weeks later.
A full description of the mRANO criteria utilized to derive the lesion response can be found at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5398984/
Evaluation of the lesion response combines the MRI scan results, clinical neurological assessment and corticosteroid doses. Additional information can be found in the Imaging Charter for the study.
Response Review
During the conduct of the study, responses will be determined using mRANO criteria by the local investigator in conjunction with a local MRI radiologist. These determinations will be used for clinical decision making regarding subjects in the study (for example, whether a subject should be withdrawn for progressive disease). All MRI scans will also be reviewed centrally by an MRI radiologist(s)at conclusion of the clinical trial.
Statistical Considerations
The study endpoints are described in Section 3. A detailed description of the analysis to be performed is described in the Statistical Analysis Plan (SAP). Therefore, a summary of the SAP is included here.
Cohorts for analysis
Full analysis set (FAS)
The FAS consists of all subjects for whom informed consent has been obtained. The FAS will be used to list and summarize patient disposition.
The safety set (SS)
The SS consists of all subjects randomized to study treatment who received at least one dose of NANO2/placebo or TMZ or radiation who also have at least one valid post-baseline safety assessment. Note that the statement that a subject that had no adverse events (as ticked on the adverse event page of the CRF) constitutes a valid safety assessment for the purposes of this study. Subjects in the safety set will be classified according to the actual treatment received (NanO2 or placebo). The SS will be used to provide listings and summaries of all safety data.
The intent-to-treat (ITT) Set
The ITT population consists of all subjects randomized to study treatment. Subjects will be analysed according to the treatment to which they were randomized. The ITT will be used to list and summarize all baseline, demographic, and efficacy information.
The per protocol set (PPS)
The PPS population consists of all subjects in the ITT set that complete at least 80% of chemoradiation treatments. For the purposes of the study, any subject that completes at least 24 of the planned 30 days of therapy in the chemoradiation period will be included in the PPS, regardless of their continued involvement in the recovery period, adjuvant TMZ period, and follow-up phases of the study.
The only exceptions to this rule will be if a subject dies during the chemoradiation period or a subject has documented disease progression during the chemoradiation period – these subjects will still be included in the PPS. The PPS will be used to perform supportive analysis of the primary efficacy endpoints, and secondary efficacy endpoints relating to ORR.
Baseline and demographic information
All demographic (age, gender, race) information will be comprehensively summarised for the ITT population by treatment group using descriptive statistics (mean, median, minimum, maximum and standard deviation) for continuous variables and frequency tables (number and percentage) for categorical variables. The stratifying variables (MGMT status and RTOG RPA class) will be summarized. In addition, IDH1 status will be summarized. Other baseline information (medical history, GBM history, medications) will be summarised by treatment group for the ITT population using frequency tables for categorical variables.
Treatments (study treatment, concomitant medications, compliance)
The total dose of NanO2 received during the chemoradiation period of the study, and the number of days on which NanO2 was administered, will be summarized using descriptive statistics.
Similar data presentations will be provided for TMZ administration in the chemoradiation period, including total dose and average daily dose. In addition, the number of cycles of TMZ started during the adjuvant TMZ period will also be presented.
The total dose of radiation received and the number of days on which radiation was administered during the chemoradiation phase will be summarized.
Concomitant medications and significant non-drug therapies prior to and after the start of the study drug will be summarized.
Efficacy analysis
General considerations
This is a phase 2 study with the aim of providing an indication of response to treatment with NanO2 compared with standard of care. Accordingly, as discussed by Rubinstein et al [30], a relaxed alpha will be used. Comparisons between NanO2 and standard of care will be conducted using a one-sided alpha of 0.2, testing the null hypothesis of no difference between treatments against the one-sided alternative, that NanO2 is superior to standard of care.
Primary efficacy endpoint
The primary efficacy endpoint is PFS. PFS will be defined as the number of days from the date of randomization to the date of the first progression-defining event: death (from any cause) or disease progression assessed using mRANO criteria. For the primary analysis the mRANO classification assigned by the central reviewer will be used. Subjects who do not progress will be censored at the date of the last assessment of disease status.
Kaplan Meier curves will be produced for PFS. Median PFS in each treatment group will be estimated. The difference in PFS between the NanO2 and standard of care treatment groups will be assessed using a stratified log-rank test, with the stratifying variables as described below.
Lifetable methods will be used to obtain estimates of PFS 6 and 12 months for each treatment group.
In addition, a cox proportional hazard model will be fitted with treatment (NanO2 or standard of care) and the stratifying variables (MGMT status, RTOG RPA, see section on randomization) as factors. IDH1 status will also be included in the model. From the model the benefit of treatment with NanO2 expressed as the hazard ratio will be estimated and presented with confidence limits.
Secondary efficacy endpoints
OS will be defined as the number of days from the date of randomization to the date of death from any cause. Subjects who do not die will be censored at the date of the last contact. OS will be analyzed similarly to PFS above.
Response assessment based on mRANO criteria as complete response (CR), partial response (PR), progressive disease (PD) or stable disease (SD). The subject’s best overall response will be determined. Subjects will be classified as responders if the best overall response is CR or PR.
The percentage of subjects in each treatment group classified as responders will be summarized.
In addition, a logistic regression model will be fitted with response Yes or No as the outcome variable and treatment group as a factor. Stratifying variables and IDH1 will be included in the model, as described for the primary endpoint. From the model the odds ratio for the odds of a response in the NanO2 group compared with the standard of care group will be estimated and presented with confidence limits.
The difference pre-treatment versus post-treatment in the TOLD MRI values will be compared between the two groups using a linear mixed effects model to account for the correlation between the measurements obtained.
FACT-Br QOL patient questionnaires will be scored according to the authors instructions to derive subscale scores, namely physical wellbeing (PWB), social wellbeing (SWB), emotional wellbeing (EWB), functional wellbeing (FWB) and the brain cancer subscale (the additional concerns, BrCs).
In addition, the following total scores will be derived:
FACTG total (PWB+EWB+SWB+FWB)
The FACT_Br total score (FACTG total + BrCs)
FACT-Br trial outcome index ( PWB+FWB+BrCs).
For each post-baseline assessment, the change from baseline (Dose 1 Chemoradiation) will be determined.
A mixed model for repeated measures (MMRM) will be fitted with change from baseline as the outcome and treatment and timepoint as factors. Stratifying variables and IDH1 will be included in the model, as described for the primary endpoint. The repeated measures nature of the data will be appropriately modelled through the inclusion of subject as a random term.
A similar approach will be followed for the CQOLC to analyze caregiver QOL.
Safety analysis
All safety summaries will be produced by treatment group using the Safety Set (SS).
Incidence of adverse events
All adverse events reported will be coded using MedDRA. The incidence of treatment emergent adverse events (new or worsening from baseline) will be summarized by system organ class and by preferred term by treatment group. Summaries will be presented overall, by severity and by the Investigator’s opinion of whether the event was related to NanO2 or placebo.
Laboratory data
All laboratory data will be comprehensively summarized by visit and treatment group.
Other safety data
Vital signs will be summarized by visit and treatment group. Physical examination findings will be listed.
Sample size
For the phase 2 component of the study randomization to NanO2 versus placebo will be in a 2:1 ratio (58 subjects randomized to NanO2 versus 29 randomized to placebo). Accrual will occur over 12 months with an additional 12 months of follow-up for PFS and an additional 36 months of follow-up for OS. This sample size will allow us to detect a 35% improvement (HR = 0.65) in PFS (OS), with 80% statistical power at a one-sided alpha level of 0. The relaxed alpha level of 0.20 was used based on the recommendation that randomized Phase 2 screening trials relax the alpha level to allow assessment of the treatment response without overly inflating the required sample size [30]. Analysis for PFS will occur when 67 PFS events (progression or death) have occurred, while the analysis for OS will occur when 67 deaths have occurred. It is expected that the PFS analysis will occur approximately 18 months after study initiation (first subject randomized), while the analysis for OS will occur approximately 42 months after study initiation. The sample size calculation was performed using SAS [31].
The median PFS for glioblastoma subjects treated with radiotherapy plus oral temozolomide in the AVAglio study was 6.2 months [32]. The proposed sample size will allow us to detect an increase in the median PFS to 9.5 months. The median survival for glioblastoma subjects treated with radiotherapy plus oral temozolomide was 14.6 months [2]. The proposed sample size will allow us to detect an increase in median survival to 22.4 months.
Results and Discussion
DATA reporting
Adverse event lists, guidelines and instructions for reporting can be found in Section 10.
Data collection
An eCRF will be utilized for data entry from source documents by research personnel at participating sites. Once data are entered, they will be systematically checked using validations programs. Queries generated by the validation programs will be displayed on the screen and via a query report. Research personnel can address the queries at any time. In addition, the independent monitor (see below) will be able to post manual queries in the eCRF for the attention of the research personnel. Any data query resolutions requiring an update to the eCRF will be performed by research personnel and all changes will be automatically added to an electronic audit trail. On completion of the study, the Principal Investigator will be required to review and electronically sign all CRFs.
Study monitoring and data queries
The Sponsor will arrange for monitoring at intervals during the study to verify adherence to the protocol and the principles of GCP, and the completeness and accuracy of the data. A monitoring plan will be prepared to describe the details of the monitoring process.
Study auditing
The investigator will permit study-related monitoring, audits, and inspections by the IRB, the study Sponsor, and government regulatory bodies all study related documents. The investigator will ensure the capability for inspections of applicable study-related facilities. Participation as an investigator in this study implies acceptance of potential inspection by government regulatory authorities and applicable compliance offices. The FDA may choose to inspect the study records as indicated in 21 CFR 312.58.
Record retention
It is the investigator’s responsibility to retain study essential documents for 2 years following the date a marketing application is approved for the drug for the indication for which it is being investigated (21 CFR 312.62). These documents should be retained for a longer period if required by an agreement with the funding entity or the supplier of the investigational agent. In such an instance, it is the responsibility of the funding entity or supplier of the investigational product to inform the investigator/institution as to when these documents no longer need to be retained.
Publication policy
Any manuscripts reporting the results of this clinical trial must be provided to NuvOx by the Principal Investigator for advisory review and comment prior to submission for publication. NuvOx will have 30 days from the date of receipt for review and will also have the right to request that publication be delayed for up to an additional 30 days in order to ensure that confidential and proprietary data, and intellectual property rights, are protected. Copies of meeting abstracts must be provided to NuvOx for courtesy review as soon as possible and at least three (3) days prior to submission. Press releases and other media presentations must also be forwarded to NuvOx prior to release.
Ethical and Reulatory Requirments
Informed consent
The investigator must obtain written informed consent from the subject after an adequate explanation of the rationale, methods, anticipated benefits and potential risks involved with the study, prior to the commencement of any study protocol-specific procedures. The informed consent process should be documented in the subject’s medical record and a copy of the signed consent form should be provided to the subject. The investigator is also responsible for notifying the subject’s primary-care physician of the participation in the clinical study if the subject agrees to such notification.
Institutional review board
A copy of the protocol and the proposed subject information and consent form must be submitted to the Institutional Review Board (IRB) for written approval at each participating site. A copy of the written approval of the protocol and consent form must be received by the clinical site before recruitment of subjects into the study and shipment of NanO2 to the site. The investigator must submit and, where necessary, obtain approval from the IRB for all subsequent protocol amendments and changes to the informed consent document. The investigator should notify the IRB of serious adverse events occurring at the site and other adverse event reports received from the Sponsor in accordance with local procedures.
Subject confidentiality
The investigator must ensure that the subject’s confidentiality is maintained. In the eCRFs or other documents submitted to the Sponsor, subjects should be identified by a subject identification number only. In compliance with regulatory requirements and ICH guidelines, the investigator and their institution will permit authorized study monitors, representatives of a regulatory agency and the IRB to review the subject’s original medical records for verification of study-related procedures and data. The investigator is obligated to inform and obtain the consent of the subject to permit this access to his/her study-related records.
Protocol amendments
The IRB and FDA must be informed of all amendments to the study protocol and the IRB must give approval prior to implementation. The investigator must send a copy of the approval letter from the IRB to the Sponsor.
Study termination
The Sponsor reserves the right to terminate the study at any time. The investigator should notify the IRB in writing of the study’s completion or early termination and send a copy of the notification to the Sponsor. Subjects may be eligible for continued treatment with NanO2 by an extension protocol or as provided for by local regulatory mechanisms.
Discussion
This trail seeks to determine if NanO2, an oxygen delivery therapeutic, can extend the PFS and OS for newly diagnosed glioblastoma patients. The phase 1 safety trail contained only 11 patients and not only was the drug considered safe at the 0.1 mL/kg dose, but it also showed a trend toward efficacy for both PFS and OS. Thus, this Phase 2 trial has been powered accordingly with an α<0.20 using 87 patients. To date, only 11 patients have been treated but the study is blinded therefore the sponsor cannot yet assess the data.
Conclusion
Among the 11 patients that have been treated, there have been no adverse effects determined to be drug related. There are currently 9 clinical sites initiated and enrolling patients. Five more sites are underway which will bring it up to a total of 14 recruiting sites. Enrollment has increased in Q2 2024 and is expected to continue increasing as the 5 new sites are brought on board. NuvOx expects to complete enrolling subjects by the middle 2025.
Abbreviations
ALT: Alanine transaminase; ANC: Absolute neutrophil count; APTT: Activated Partial Thromboplastin Time; AST: Aspartate transaminase; BOLD: Blood Oxygenation Level Dependent MRI; BP: Blood Pressure; CNS: Central Nervous System; CO: Carbon Dioxide; CQOLC: Caregiver Quality of Life – Cancer; CRF: Case Report Form; CSF: cerebrospinal fluid; CTCAE: NCI Common Terminology Criteria for Adverse Events, Version 5.0; CTV: Clinical Target Volume; DDFP: Dodecafluoropentane; DICOM: Digital Imaging and Communications in Medicine; DLT: Dose Limiting Toxicity; DNA: Deoxyribonucleic acid; eCRF: Electronic Case Report Form; EDC: Electronic Data Capture System; FACT-Br: Functional Assessment of Cancer Therapy-Brain; GBM: Glioblastoma Multiforme; GCP: Good Clinical Practice; Gd-MRI: Gadolinium-enhanced MRI; GTV: Gross Tumor Volume; Gy: Gray; HED: Human Equivalent Dose; HEENT: Head, Eyes, Ears, Nose, and Throat; IDH1: Isocitrate dehydrogenase 1; IMRT: Intensity-Modulated Radiation Therapy; INR: International normalized ratio; IRB: Institutional Review Board; IV: Intravenously; kg: Kilograms; KPS: Karnofsky Performance Status; LDH: Lactate dehydrogenase; LTFU: Long-term Follow Up; MCV: Mean cell volume; MCH: Mean corpuscular hemoglobin; MCHC: Mean corpuscular hemoglobin concentration; MGMT: Methylguanine methyltransferase gene; MI: Myocardial Infarction; MIDSS: Measurement Instrument Database for the Social Sciences; mRANO: Modified Response Assessment in Neuro-oncology; MRI: Magnetic Resonance Imaging; MTD: Maximum Tolerated Dose; NCI: National Cancer Institute; NYHA: New York Heart Association; OS: Overall Survival; PCP: Pneumocystis Pneumonia; PD: Pharmacodynamic; PFS: Progression-Free Survival; PICC: Peripherally-Inserted Central Catheter; PK: Pharmacokinetic; PTB: PolyEthyleneGlycol- Telomer B; RD: Recommended Dose; RT: Radiation Therapy; RTOG-RPA: Radiation Therapy Oncology Group Recursive Partitioning Analysis; SAE: Serious Adverse Events; SpO2: Oxygen saturation by pulse oximetry; SUSAR: Serious and unexpected suspected adverse reaction; TEAE: Treatment Emergent Adverse Event; TMZ: Temozolomide; TOLD: Tissue Oxygenation Level Dependent MRI; TSC: Trans Sodium Crocetinate; ULN: Upper Limit of Normal.
APPENDICIES
Appendix A: REFERENCES
Appendix B: Karnofsky Performance Status
Appendix C: Schedule of Events
Appendix D: FACT-Br Questionnaire
Appendix E: CQOLC Questionnaire
Acknowledgement
Protocol Number: RESTORE P2
Version and date: Version 11.0 - 21 November 2022
Sponsor: NuvOx Pharma, 1635 East 18th Street, Tucson, Arizona, USA 85719
ClinicalTrials.gov Identifier: NCT03862430
References
1. Dunn G, Rinne M, Wykosky J. Emerging insights into the molecular and cellular basis of glioblastoma. 2012;26:756–84.
2. Stupp R, Mason W, van den Bent M. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. 2005;352:987–96.
3. Butowski N, Sneed P, Chang S. Diagnosis and treatment of recurrent high-grade astrocytoma. 2006;24:1273–80.
4. Francesconi A, Dupre S, Matos M, Martin D, Hughes B, Wyld D, et al. Carboplatin and etoposide combined with bevacizumab for the treatment of recurrent glioblastoma multiforme. 17:970–4
5. Bluff J. Tissue factor, angiogenesis and tumour progression. 2008;10(2):204.
6. Ogiichi T, et al. Tissue factor and cancer procoagulant expressed by glioma cells participate in their thrombin-mediated proliferation. 2000;46(1):1–9.
7. Yao XH, et al. Production of angiogenic factors by human glioblastoma cells following activation of the G-protein coupled formylpeptide receptor FPR. 2008;86(1):47–53.
8. Wachsberger P, Burd R, Dicker A. Improving tumor response to radiotherapy by targeting angiogenesis signaling pathways. 2004;18(5):1039–57.
9. Peles E, Lidar Z, Simon A. Neurosurgery. 2004;55(3):562–8.
10. Maity A, Pore N, Lee J. Cancer Res. 2000;60:5879–86.
11. Gray L, et al. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. 1953;26(312):638–48.
12. Bristow RG, Hill RP. Hypoxia and Metabolism: Hypoxia, DNA Repair and Genetic Instability. 2008;8(3):180–92.
13. Rockwell S, Dobrucki I, Kim E, Marrison S, Vu V. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. 2009;9(4):442–58.
14. Sun S, Lee D, Lee N. Hyperoxia resensitizes chemoresistant human glioblastoma cells to temozolomide. 2012;109(3):467–75.
15. Baddeley H, Brodrick P, Taylor N, et al. Gas exchange parameters in radiotherapy patients during breathing of 2%, 3.5% and 5% carbogen gas mixtures. 2000;73:1100–4.
16. Miralbell R, et al. Accelerated radiotherapy, carbogen and nicotinamide in glioblastoma multiforme: report of European Organization for Research and Treatment of Cancer trial 22933. 1999;17:3143–9.
17. Kokunai T, Kuwamura K. Effect of Perfluorochemicals on BCNU Chemotherapy: Preliminary Study in a Rat Brain Tumor Model. 1982;18(4):258–61.
18. The Effects of NVX-108 as a Radiation Sensitizer in Glioblastoma. Available from: https://clinicaltrials.gov/ct2/results?term=nuvox&Search=Search
19. Rose C. Int. J. Radiation Oncology Biol Phys. 1986.
20. Evans R, et al. Int. J. Radiation Oncology Biol Phys. 1990.
21. Hochberg F. Journal of Neuro-Oncology. 1997.
22. Dowling S. Fluosol and hyperbaric oxygen as an adjunct to radiation therapy in the treatment of malignant gliomas: a pilot study. 1992;903–5.
23. Johnson J, et al. Radiation sensitization of Hs-766T pancreatic tumor xenografts in mice dosed with dodecafluoropentane nano-emulsion.
24. Koch CJ, Oprysko PR, Shuman AL, Jenkins WT, Brandt G, Evans SM. Radiosensitization of Hypoxic Tumor Cells by Dodecafluoropentane A Gas‐Phase Perfluorochemical Emulsion. 2002;62(13):3626‐3629.
25. Schafer R, Gmitro AF. Dynamic Oxygenation Measurements Using a Phosphorescent Coating within a Mammary Window Chamber Mouse Model. 2016Dec14;6(2):639–50.
26. Reigner B, Blesch K. Estimating the starting dose for entry into humans: principles and practice. 2002;57(12):835–45.
27. USDoHaH S, FaD A, CfDEaR (CDER). Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005.
28. Available from: https://www.facit.org/FACITOrg/Questionnaires
29. Weitzner MJ, Wagner H, Friedland J, Cox C. The Caregiver Quality of Life Index-Cancer (CQOLC) scale. 1999. Available from: http://www.midss.org/content/caregiver-quality-life-index-cancer-cqolc-scale
30. Rubinstein L, Korn E, Freidlin B, Hunsberger S, Percy Ivy S, Smith M. Design Issues of Randomized Phase II Trials and a Proposal for Phase II Screening Trials. 2005;23:7199–206.
31. 15 Power Analysis and Sample Size Software. 2017.
32. Chinot O, Wick, Mason, Henriksson, Saran, Nishikawa, et al. Bevacizumab Bevacizumab plus-Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. 2014;370(709-22).
33. Fontanilles M, Marguet F, Alexandru C, Langlois O, Veresezan O, Gilard V, et al. Early platelet variation during concomitant chemo-radiotherapy predicts adjuvant temozolomide-induced thrombocytopenia in newly diagnosed glioblastoma patients. 2019;27(2):477–84.
34. Ellingson B, Wen P, Cloughesy T. Modified Criteria for Radiographic Response Assessment in Glioblastoma Clinical Trials. 2017;14(2):307–20.
35. Kitzman D, Wesley D. Safety assessment of perflenapent emulsion for echocardiographic contrast enhancement in patients with congestive heart failure or chronic obstructive pulmonary disease. 2000Jun26;139(6):1077–80.
Received: May 1, 2024;
Accepted: June 3, 2024;
Published: June 10, 2024.
To cite this article : LH Johnson J, Unger E. A phase 2 Double-Blind, RandomizEd, Prospective, Placebo Controlled Study of NanO2TM Combined With Radiation and Temozolomide in Patients with Newly-Diagnosed Glioblastoma MultiformE: RESTORE. British Journal of Cancer Research. 2024; 7(2): 694- 715. doi: 10.31488/bjcr.195.
© The Author(s) 2024. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/).